CATALYSIS RESEARCH
1. Mechanistic Study of Electrocatalytic Perchlorate Reduction using an Oxorhenium Complex Supported on a Ti4O7 Support

Schematic of perchlorate reduction on Re/Ti4O7 electrode. (a) ClO4– adsorption; (b) OAT from ClO4– to Re/Ti; (c) ClO3– reduction at reduced Ti sites; and (d) regeneration of Re(VII) to Re(V) via proton reduction. Adapted Image from our publication in ACS Catal. 2024, 14, 2597– 2608
Developing a stable and active catalyst for ClO4– reduction at nonacidic pH has presented a significant challenge to the catalysis field. Previous research has demonstrated that by depositing an organometallic Re catalyst onto a Ti4O7 support (Re/Ti4O7), it was possible to stabilize the catalyst and obtain active electrocatalytic ClO4– reduction at circumneutral pH. Thus, the focus of this work, in collaboration with Prof. Chaplin et. al., UIC (experiment lab) was on elucidating the mechanisms of electrocatalytic ClO4– reduction in water with the Re/Ti4O7 system. Density functional theory (DFT) simulations indicated that the adsorption of the Re catalyst was exothermic on Ti4O7, and X-ray photoelectron spectroscopy (XPS) characterization indicated that Re adsorption caused a net reduction of the Ti oxidation state on the Ti4O7 surface. After ClO4– reduction experiments, XPS results indicated the presence of Ti(0)/Ti(II) surface sites. Cyclic voltammetry experiments in an acetonitrile solvent provided supporting evidence that these surface sites were electroactive and likely participated in the ClO4– reduction reaction. Analysis of batch reduction experiments in acetonitrile via kinetic modeling estimated a catalyst turnover number of 332 ± 23, which provided further evidence that the reduced Ti sites could regenerate the Re catalyst. However, these reduced Ti sites were finite in number and required the production of adsorbed hydrogen via water reduction to facilitate continuous ClO4– reduction. DFT results indicated that the reduction of ClO4– to Cl– was exothermic and that reduced Ti sites participated in the reduction reaction. The experimental and DFT results allowed a preliminary mechanism for ClO4– reduction on Re/Ti4O7 to be proposed.
Reaction profile for implicit water solvation model

Reaction profile for implicit water solvation model. In this case, the decomposition of ClO4– on the [ReO(hoz)2]+ adsorbed TiO7 surface is exothermic with −10.4 eV of energy. Reactions that release Cl– are more exothermic (i.e., −15.4 eV). Adapted Image from our publication in ACS Catal. 2024, 14, 2597– 2608
The findings of this study enhanced our understanding of the key processes involved in the electrocatalytic reduction of ClO4– in water by using the Re/Ti4O7 electrode. The DFT simulations revealed that the Re catalyst exhibited exothermic adsorption on Ti4O7 and remained stable in both the Re(V) and Re(VII) oxidation states. Experimental studies were performed by Dr. Chaplin Group.
2. Sequential Hydrolysis of Metal Oxo Clusters in Zirconium Metal–Organic Frameworks
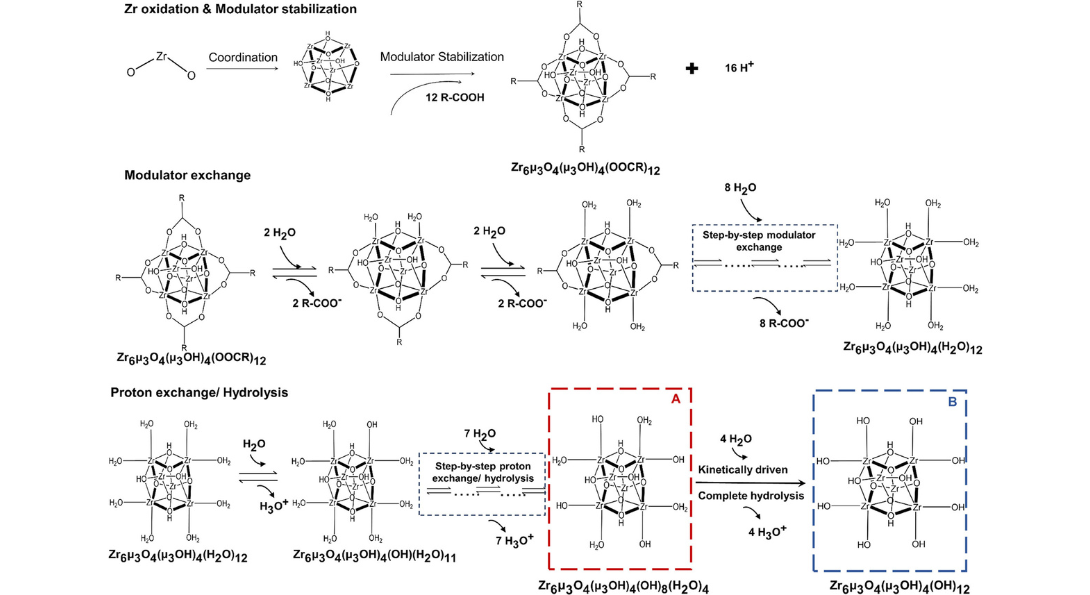
Zr-oxo Cluster Evolution. Adapted from our publication in Nanoscale 2023, 15, 9329– 9338, DOI: 10.1039/D2NR06685H
A detailed deprotonation mechanism of the evolution of Zirconium (Zr) oxo cluster directing the synthesis of porphyrin-based zirconium MOFs is presented. The density functional theory calculations reveal that zirconium-oxo clusters undergo sequential hydrolysis, with the pKa of the cluster dictating the extent of deprotonation. The degree of deprotonation, in turn, determines the 12- and 8-connections in MOF-525 and PCN-222, respectively.
Experimental studies were performed by Dr. Meenesh Singh Group.
3. Electrochemical Reactions Profile on the Cathode Surface in Li-CO2 Batteries

A) Density of state calculations for Sb0.67Bi1.33Te3 and Sb0.67Bi1.33S3. B) Free energies of steps in the CO2 reduction reaction pathway on Sb0.67Bi1.33Te3. C) Sb (110) surface. D) CO2 on surface. E) LiCO2 on surface. F) Li2CO2 on surface. G) C-CO3 on surface. H) Schematic of the Li+-electron coupling reaction to form LiCO2. I,J) AIMD results for CO2 in EMIM-BF4/DMSO/1 m LiTFSI electrolyte with: I) 6:4 and J) 9:1 volume ratio of DMSO/EMIM-BF4. Adpated from our publication inAdvanced Energy Materials 2024, 14, 2303467.
In this study, we investigate two novel transition metal trichalcogenide (TMTC) alloy catalysts, Sb₀.₆₇Bi₁.₃₃X₃ (X = S, Te), for application in Li–CO₂ batteries. Leveraging density functional theory (DFT) as a central tool, we explore the underlying electronic and catalytic properties of these materials and their interactions with ionic liquid-based electrolytes. These theoretical insights are complemented by experimental validation through a collaboration with the Amin Salehi group.
Our DFT analyses reveal that the choice of chalcogen (Te vs S) significantly impacts the electronic structure of the catalysts. Specifically, the Te-based compound exhibits a smaller bandgap and enhanced CO₂ reduction (CO₂RR) and evolution (CO₂ER) activity compared to its S-based counterpart, explaining its superior performance in catalytic cycles. These predictions are corroborated by electrochemical testing, where the Te-based catalyst demonstrates exceptional stability and activity—sustaining a current density of 1 mA cm⁻² over 220 cycles.
Furthermore, our simulations suggest that CO₂RR is driven by a coupled lithium cation–electron transfer mechanism, offering insight into the reaction pathway and catalytic efficiency. The combination of favorable electronic structure, tunable electrolyte interface, and robust alloy stability underscores the potential of Sb₀.₆₇Bi₁.₃₃Te₃ as a high-performance catalyst for Li–CO₂ batteries.
This work highlights how DFT-guided design enables the rational development of next-generation catalysts capable of sustaining high current densities—addressing one of the key bottlenecks in CO₂ battery technologies.
Published in: Advanced Energy Materials 2024, 14, 2303467.
4. Lithium superoxide-based high rate Li-Air batteries enabled by Di-iridium sulfur bridge active sites
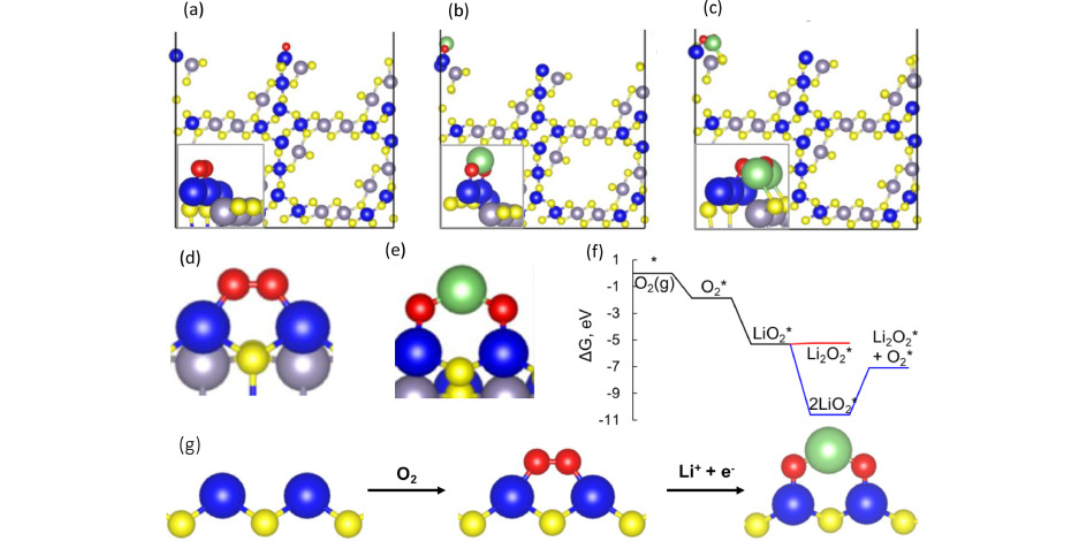
Adapted figure from Energy Storage Materials 60 (2023) 102844. The most favorable adsorption configurations of (a) O2, (b) LiO2, and (c) Li2O2 on the Ir terminated IrSnS3.6(100) surface. Close-up structures of O2 in (a) on the di-Iridium sulfur bridge site are shown in (d) and LiO2 in (b) is shown in (e) from a different angle. Reaction free energies (f) plot for LiO2 formation and Li2O2 formation on the Ir terminated IrSnS3.6(100) surface. The Ir, Sn, S, O, and Li atoms are in blue, light purple, yellow, red, and green, respectively. Black lines in (a)–(c) are the periodic boundaries. The blue lines in (f) represent the LiO2 disproportionation reaction pathway, while the red lines show a (Li+ + e−) transfer to LiO2 to form Li2O2. (g) Schematic illustrating the involvement of the di-iridium sulfur bridge site in the LiO2 reaction. (For interpretation of the references to color in this figure, the reader is referred to the web version of this article)
Density Functional Theory (DFT) calculations revealed that the di-Ir–S bridge site on the Ir-terminated SnIrS₃.₆(100) surface is the most favorable active site for O₂, LiO₂, and Li₂O₂ adsorption. DFT-derived reaction free energies confirmed that LiO₂ prefers reduction (via Li⁺ + e⁻) to form Li₂O₂ rather than disproportionation, aligning with observed discharge behavior. The calculated adsorption geometries and charge distribution support the enhanced reversibility and catalytic efficiency seen experimentally by our collaborators lead by Dr. Salehi. These insights rationalize the low overpotentials and robust cycling performance of SnIrS₃.₆ in Li–O₂ batteries.
Published in: Energy Storage Materials 2023, 60, 102844.