OUR DFT SIMULATION CAPABILITIES
Introduction
Various theoretical methods such as Density Functional Theory (DFT), Time-Dependent DFT (TDDFT), Ab Initio Molecular Dynamics (AIMD), and Multiconfigurational methods (MC-PDFT, CASSCF, CASPT2) are used for quantum chemistry calculations.
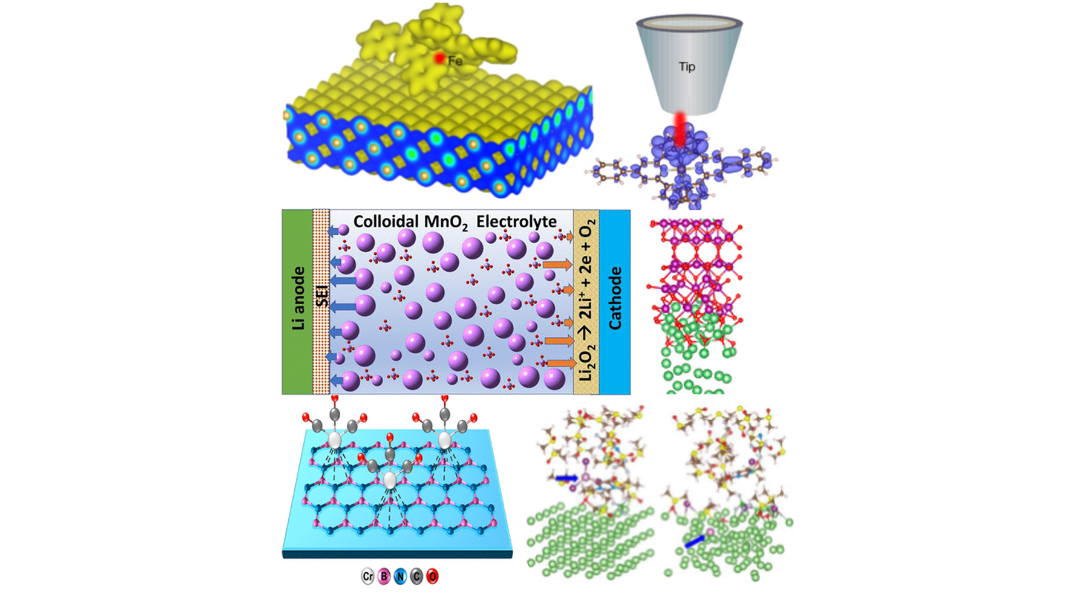
- DFT calculations were used in validating Fe oxidation states and local coordination and density of states that match with experimental characterization using synchrotron X-Rays.
- DFT computations explained adsorbed coordination of Cr(CO)x on Boron Nitride nanosheets.
- Our AIMD/DFT calculations provided insights to High-Rate Long Cycle-Life Li-Air Battery in presence of redox mediators.
- We conduct surface energy, adsorption, and reaction energy calculations to understand electronic structure, catalyst behavior, CO₂ reduction, and electrolyte interactions in Li and beyond Li batteries and energy storage systems.
Selected Publications
- Nature 618, 69–73 (2023)
- ACS Nano 2022, 16, 11, 18187–18199
- ACS Appl. Nano Mater. 2025, 8, 1, 233–243
- ACS Appl. Mater. Interfaces 2021, 13, 4, 4915–4922